
Insider Brief
- A Cleveland Clinic–IBM team demonstrated a hybrid quantum-classical workflow to approximate the electronic structure of the 303-atom Trp-cage protein using IBM Quantum Heron r2.
- The workflow combines wave function-based embedding to fragment the protein into clusters and sample-based quantum diagonalization to solve complex electronic structures.
- The approach scales beyond Trp-cage and could support pharmaceutical research and molecular simulations using quantum-centric supercomputing.
A joint Cleveland Clinic-IBM research team has used quantum computing to demonstrate a hybrid quantum-classical workflow to approximate the electronic structure of a protein for the first time, IBM announced. The team modeled the 303-atom miniprotein Trp-cage using a quantum-centric supercomputing workflow and an IBM Quantum Heron r2.
The work represents progress in designing and applying quantum-centric supercomputing algorithms and workflows that combine quantum and high-performance classical computing.
Accurate electronic structure calculations on classical computers become increasingly challenging as system size increases, the company stated. Classical methods alone can efficiently model certain aspects of protein behavior, but high-accuracy quantum-mechanical treatments of entire proteins remain impractical.
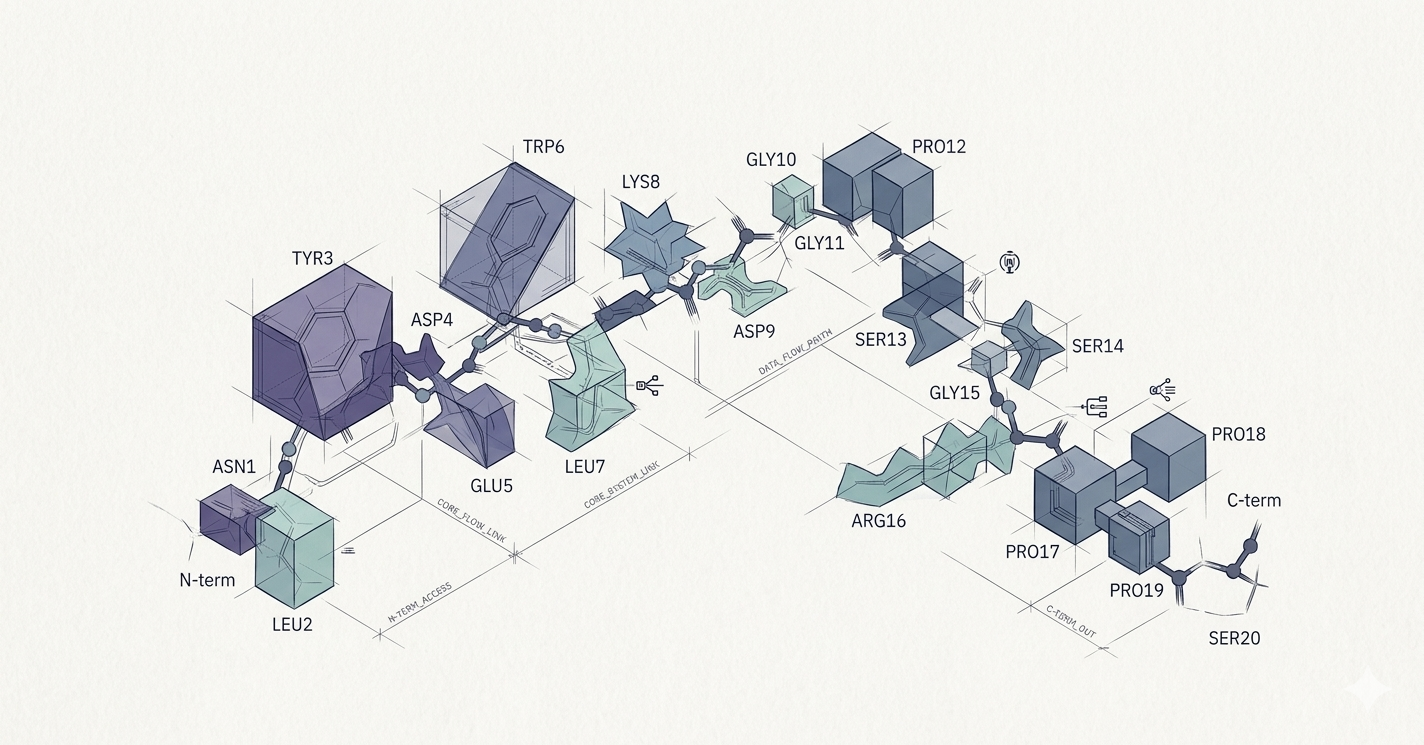
Modeling Trp-Cage Miniprotein
Trp-cage is useful for benchmarking computational chemistry methods. The molecule is relatively compact for a protein, but it has features that are common to much larger molecules in biochemistry, such as a hydrophobic core and hydrogen bonding between its constituent parts, allowing it to take on more complex structures. The researchers modeled both its unfolded and folded states.
“Proving that this approach works for Trp-cage is a step toward larger molecules,” said Mario Motta, co-author of the paper, as quoted by IBM.
The team initially planned to simulate just a couple of amino acids, Motta stated. But as they tested their workflow, they found they were able to scale all the way up to Trp-cage and get meaningful results.
Dr. Kenneth Merz, who leads the Merz lab at Cleveland Clinic, said he hopes these methods could support computational workflows for pharmaceutical research and related fields as they mature and scale. He envisions a world where scientists use quantum-centric supercomputing workflows to build databases of simulated molecular behavior, then use machine learning algorithms trained on those databases to identify molecules that might behave in needed ways.
Wave Function-Based Embedding Workflow
The workflow, described in a recent preprint on arXiv, relies on a technique called wave function-based embedding to fragment Trp-cage into computationally tractable pieces called “clusters,” source reported. In this approach, there are as many clusters as there are atoms in the molecule, but each cluster is more complex than a single atom, encompassing a local region surrounding the atom and entangled with it.
Some clusters are much more complex than others, the company explained. One atom may be at the edge of the protein and only entangled to one or two neighboring atoms, allowing researchers to find that cluster’s electronic structure efficiently using classical computational methods. Another might be closer to the molecular core, enmeshed in a more complex web of intermolecular interactions. These larger clusters are good problems for quantum computers to solve.
Stitched back together, the results of individual cluster calculations lead to a complete solution for the electronic structure of the molecule, which describes where its electrons are and how they interact.
Sample-Based Quantum Diagonalization Algorithm
Merz has watched the development of quantum computing over several years, according to IBM. Until a few years ago, it was clear that quantum computers could offer new approaches to solving hard chemistry problems, but what those approaches would look like remained an open question.
Merz said there was a eureka moment when he saw IBM scientists present an algorithm called sample-based quantum diagonalization (SQD). The algorithm belongs to an emerging set of algorithms built for quantum-centric supercomputing, where classical and quantum resources work together to solve problems using the strengths of both paradigms, as reported by IBM.
Sample-based quantum diagonalization (SQD) addresses one of the fundamental challenges of electronic structure calculations – the number of possible configurations of a molecule’s electrons grows combinatorially with the molecule’s size. In the algorithm, the quantum computer samples this vast space, identifying key configurations for the classical computer to focus on. The classical computer uses the resulting information to find a solution.
“We sort of dropped everything. I met with a few people in my group over the weekend, and we decided to just go all in on SQD,” Merz stated.
The team began testing the algorithm on a string of smaller molecules, beginning a chain of experiments that led to this Trp-cage simulation. The results so far have been extremely promising, with the workflow already performing competitively with classical approaches and approaching the accuracy of the most computationally demanding among them, source reported.
In principle, the combined wave function-based embedding and sample-based quantum diagonalization workflow can scale far beyond Trp-cage, the scientists said. As molecules get larger, the task of breaking them up, calculating their most complex clusters, and stitching them back together gets more complex. But solving for the electronic structure of complex clusters is a compelling problem for quantum computers. The researchers are exploring what the next step looks like, eyeing even larger molecules as their targets.
The work was made possible by access to high-performance computing resources at Michigan State University and Cleveland Clinic, IBM stated. Other recent collaborations between IBM and HPC leaders like RIKEN have also yielded results.
Source — Cleveland Clinic and IBM debut new quantum workflow for simulating proteins.