

Insider Brief
- A recent study by a multi-institutional team of researchers presents a new approach to solving chemical simulation problems using quantum computing.
- The work could lead to more efficient computational chemistry that could, in turn, lead to faster drug development and materials innovation.
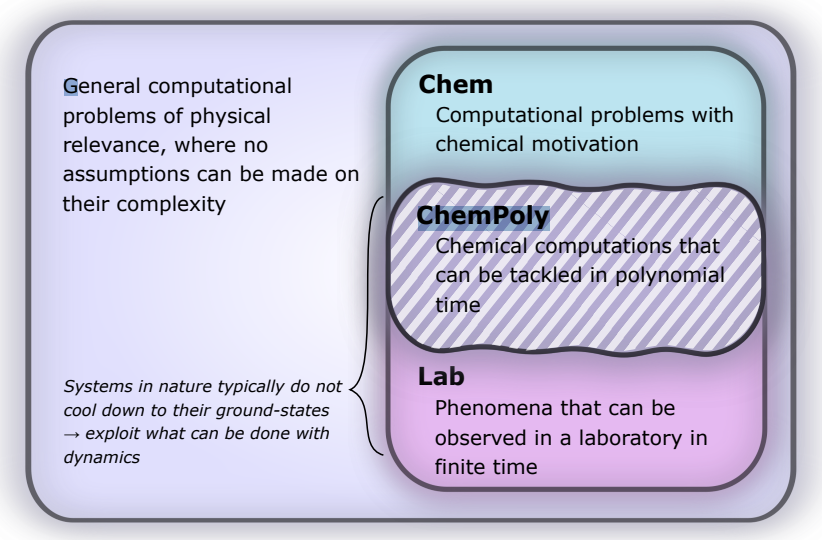
- Image: Complexity of simulating chemical problems. We target the set of computational problems, ChemPoly, within chemistry that have polynomial complexity when simulated on a quantum computer and are observable in a lab; the sets Lab and ChemPoly may coincide. (From the study)
A recent study by a multi-institutional team of researchers presents a new approach to solving chemical simulation problems using quantum computing, potentially offering a more efficient pathway in computational chemistry. The study, published recently in the pre-print server ArXiv, proposes an innovative method that could significantly enhance the way chemical systems are simulated, addressing the longstanding issue of computational cost escalation with increasing system size.
The team writes: “With fault-tolerant quantum computers, dynamical simulation of quantities that a practicing chemist might directly care about is within reach – most relevant quantum chemistry problems are inherently addressable through dynamical evolution alone, leading to efficient quantum algorithms for these problems.”
The University of Toronto-led team reports that their research is a departure from traditional chemical simulations, which have been limited by the constraints of classical computing. The study builds upon foundational ideas first proposed by pioneers, including Paul Benioff, Richard Feynman, and Yuri Manin, who envisioned the use of quantum computers for simulating quantum systems.
De-Hexing The ‘Curse of Dimensionality’
The core of this research lies in an approach that bypasses the extensive computational demand typically required for determining the ground states of chemical systems. This task has traditionally been a major challenge due to the “curse of dimensionality” and has been a significant part of supercomputing usage. Simply — and probably over-simply — stated, the curse of dimensionality refers to how the complexity and computational resource requirements of an algorithm increase exponentially with the number of dimensions in the data.
The new method, however, focuses on dynamic simulations of chemical reactions, leveraging the capabilities of fault-tolerant quantum computers.
Quantum simulations, as described in the study, involve the time evolution according to the Schrödinger equation under a Hamiltonian. These simulations are within the reach of quantum computers, classified under the computational complexity class BQP (bounded error quantum polynomial time). This classification suggests that such simulations can be solved with polynomial time complexity and bounded error probability on a quantum computer.
The researchers report that their framework relies on a set of configurable atomic initial states, which are then built into input states for a reaction through a dynamic scattering process. This process, enhanced by artificial potentials, leads to the production of a molecular input state for subsequent dynamical simulations. The approach is different from previous efforts, specifically it circumvents the challenges associated with the QMA-hardness of ground-state preparation and the associated orthogonality catastrophe – the reduced probability of successfully retrieving a ground-state, the researchers write.
This new methodology promises to facilitate the simulation of a wide array of chemically relevant problems efficiently. By focusing on dynamical simulations, the researchers provide an alternative to the computationally intensive ground-state search, traditionally seen as a bottleneck in chemical simulations.
While the study lays a robust theoretical groundwork, the team acknowledges the need for further research to evaluate the practical aspects of their approach. Future investigations might include integrating classical simulation tools like the Nosé-Hoover thermostat and developing quantum heuristics for ground states. It’s also important to note that ArXiv is technically not peer-reviewed, an important step in the scientific process.
Barring intractable hang-ups uncovered in future investigations, the potential impact of this development could be substantial. By enabling more efficient simulations of chemical systems on quantum computers, the research could accelerate several different fields, including drug development, material science,and beyond. At the very least, this study highlights the evolving role of quantum computing in scientific research, as well as offers a way to advance the field of computational chemistry.
The research was led by Philipp Schleich and includes two names that should be familiar in the quantum computing industry Joe Fitzsimons, of Horizon Quantum Computing, and Alán Aspuru-Guzik, a a professor of chemistry, computer science, chemical engineering and materials science at the University of Toronto. The team also includes Lasse Bjørn Kristensen, Davide Avagliano, Mohsen Bagherimehrab, Abdulrahman Aldossary, Jorge A. Campos Angulo and Christoph Gorgulla.
For more market insights, check out our latest quantum computing news here.




